
To Cure T1D, Start with the Easiest Cases, Not the Hardest
Not all T1Ds are autoimmune, and that changes everything.


Building things usually works best when you start with the foundation and build upward. When the problem is complex, science adds a second rule: start with the simplest version of the problem, test it, learn from it, and then advance by controlled increments of complexity. That is how we built the pyramids, learned to fly, transplanted organs, and made machines that now fit in our pockets. Start with the base case. Prove what works. Then climb.
So why aren’t we applying that same logic to the quest to cure T1D?
A cure for T1D is one of the most technically complex problems in medicine because it requires solving several hard problems at once: making new insulin-producing islets, and preventing the immune system from destroying them. The field has made extraordinary progress on the first problem. The hard part now is the immune system.
For a review of these technologies and approaches, the ADA published an article in 2025 titled, Advances in Cell Replacement Therapies for Diabetes. Seventeen luminaries in the field explain the actual technologies associated with each of these challenges. Authors include James Shapiro (who developed the Edmonton Protocol, the first islet transplant in 2000), Camillo Ricordi, Bernhard Hering, Michael Rickels, and most of the others who built the field of clinical islet transplantation.
The bulk of the article explains each of four different approaches currently under investigation to tackling the immune problem:
wrap the islets in a membrane the immune system can’t cross (encapsulation)
place them where the immune system patrols least (immune-privileged sites)
edit the cells so the immune system can’t recognize them (hypoimmune islets)
retrain the immune system itself to stand down (tolerance induction)
This is why the authors claimed the immune system as “the next frontier”. And they’re right. It’s the top of the pyramid. If that frontier is crossed, the rest of the cure begins to look achievable.
But what they don’t discuss is how to get there. The current technologies are being built for the hardest cases: patients whose immune systems are fully armed, fully autoimmune, and ready to attack anything that looks like a new beta cell. By engineering against maximum hostility first, the field is trying to place the capstone before proving the foundation beneath it.
But that might not be the best way to solve the problem.
In the section of the review article dealing with stem-cell grown islets, the authors plant the idea of growing islets from the patient’s own tissues — autologous stem-cell grown islets — and just putting them back into the patient. No encapsulation, no immunosuppressants, no gene-editing. Just a straightforward procedure. They said:
“The autologous concept theoretically allows for successful transplantation without need for immunosuppression in patients without autoimmunity, substantially altering the risk-benefit for patients with diabetes in favor of transplantation.”
This would easily go unappreciated because of its simplicity. And naturally, this would only work for patients that have no autoimmunity.
Wait, T1D without autoimmunity?
Yes, that fact slides by quickly, and it’s a fact about T1D that few are aware of. Consider this striking statistic: Roughly 22% of T1Ds don’t even have autoimmunity. Accordingly, those patients don’t need most of the technologies being developed to evade the immune system.
The review article doesn’t talk about that—they only mention it because they wrote about two trials that attempted it. These trials involved involving making autologous SC-islets and putting them back into patients. The results: ideal glycemic control and the islets survived.
But the problem was that the subjects were already on immunosuppression (from organ transplant surgeries), and the patients were not actually determined to be non-autoimmune. The trials were essentially an attempt just to see if autologous islets would work. They did.
The paper then moves on.
But, let’s pause and consider that autologous SC-islet idea more closely. This isn’t just a curiosity — when we dive into the science and the medical literature, we find there’s a great deal more to this idea. And the more you look, the more that’s revealed.
This is the base of the pyramid. The easy cases that have never, ever been tested.
Why not? Two reasons, the first of which comes from the surprise you’re feeling about that 22% figure — one that is derived from multiple sources and based on testing criteria that is inconsistently applied (which we’ll dive into later). Most people think that non-autoimmune T1D is both small in number, and as statistical and medical anomalies. Freaks of nature, whose reason for even acquiring the disease could be due to any number of causes that might also have to be dealt with that may only complicate things further. This is why this subgroup is formally called, “idiopathic diabetes” — meaning, “we don’t know.”
The second reason follows closely: If there’s not that many, and they’re medical anomalies, where are the institutional incentives, historical precedents, funding sources? You never hear BreakthoughT1D or the Helmsley Trust talk about them.
But non-autoimmune T1Ds are not outliers at all for reasons that go beyond just that 22% — they’re just the base case of the autoimmunity pyramid. As we use tools to measure actual immune system biomarkers—not just historical markers like the presence of antibodies that happened long ago at onset—a more complex picture emerges: The severity and intensity of even the traditional autoimmune response is not binary at all. People’s current autoimmunity state isn’t static. The severity ladder increases, climbing up the pyramid.
Only when you get to the very top—the capstone—do you see the population with the most aggressive immune response, typically the ones who are within 5-10 years of diagnosis. And, of course, the stages 1 and 2 T1Ds, whose autoimmune systems are just starting to ramp up.
And ironically, the approach has been to focus on those fewest, hardest, and more expensive population. The ones with maximum immune response.
These degrees of immune response can be leveraged if approached strategically. For example, there’s medical literature that suggests that even many traditionally autoimmune T1Ds might be “functionally non-autoimmune” insofar as being eligible for an autologous SC-islet transplant with no immunosuppressants. LADA patients, for example.
We also find medical literature rife with those who develop “operational tolerance”, where their immune system eventually just accepts new foreign tissues. Whole organ transplant patients are common examples.
We’re spending a great deal of money, time, and complex technologies to address the maximally hostile immune system, while there’s a large proportion of the T1D population that just doesn’t need that much.
But there’s a stronger scientific reason to start with the non-autoimmune T1Ds. It is more aligned with the scientific method: start with the simplest case, remove all the confounders, isolate conditions to their most rudimentary components, and see what happens. This allows for problems to be solved one by one as we work through the complex biology of islet placement, types of immune responses, and more. Let each new discovery inform the next tier of complexity as we move up the immune-severity ladder. Along the way, we may develop curative protocols that can be applied today to certain populations whose immune responses can tolerate them.
Early trials expose failure modes, laying the foundational work needed to solve the rest of the immune system challenges as we climb the pyramid.
This article explores this approach in these discrete steps:
What non-autoimmune T1D actually is, how to identify it, and why the immune system turns out to be a spectrum rather than a switch — including how large that population really is. (Two Types, Determining f-T1B, and the Numbers section)
What the existing trials and literature already show — and exactly where they stop short of the experiment that matters. (Autologous vs. Hypoimmune)
The economic consideration — not just in the cost of trials, but the savings in R&D and other costs that can be avoided (An Economic Consideration)
We begin by understanding more about non-autoimmune Type 1 Diabetes.
Two Types of T1D and the Meandering Dividing Line
The American Diabetes Association has long divided Type 1 diabetes into two subtypes. Type 1A (T1A) is the autoimmune form — the kind nearly everyone means when they say “Type 1.” It is marked by detectable autoantibodies against the beta cell’s own proteins (most commonly GAD65, IA-2, ZnT8, and insulin itself), by high-risk HLA genes, and by the full cellular machinery of autoimmunity: T cells, B cells, and antigen-presenting cells coordinated against the patient’s own islets.
Type 1B (T1B) is the idiopathic form. The patient is insulin-dependent and ketosis-prone like any Type 1, but shows no sign of autoimmunity — no antibodies, no autoreactive markers. The beta cells were destroyed by something other than the immune system. “Idiopathic” is the formal name, and it carries its usual medical meaning: we don’t know why.
True idiopathic T1B is a diagnosis of exclusion — no autoimmunity, and no other cause anyone can name. But it sits within a wider family of non-autoimmune, insulin-deficient diabetes: cases in which the beta cells were lost to an identifiable, non-immune process. Most of these are formally classified elsewhere — often as “other specific types” — but they share idiopathic T1B’s one defining feature, the absence of autoimmunity. They include:
direct viral destruction of beta cells (as opposed to a virus that triggers autoimmunity)
toxins or drugs that are directly toxic to beta cells
diseases of the exocrine pancreas — acute or chronic pancreatitis, cystic-fibrosis-related diabetes (CFRD), and the iron overload of hemochromatosis — sometimes grouped together as “Type 3c”
and the truly idiopathic cases: the wastebasket where, by definition, no one knows
This may feel like a true dividing line, but here’s where it starts getting complicated. There are two additional causes that often get swept into this group, which include:
Diabetes from molecular mimicry, where a virus resembles a beta-cell protein closely enough that the immune system attacks both.
Diabetes triggered by cancer checkpoint inhibitors, drugs that work by releasing the immune system’s brakes.
These two sit awkwardly in the T1B camp because neither is non-autoimmune, and neither belongs on the first list above. That is, there was an autoimmune process that killed the beta cells, but it was not genetically driven. The difference is that genetic autoimmunity is durable and self-renewing — it will destroy fresh beta cells even when those cells are the patient’s own, as identical-twin transplants showed decades ago in Sutherland, Sibley, Xu et al.
By contrast, a one-time, externally triggered attack — such as molecular mimicry and checkpoint inhibitors — in someone without that genetic program may not persist at all. Same etiological bucket, opposite implications for a transplant — which is the first sign that the cause of T1D is the wrong axis to be sorting on.
Remember, the question we care about is not how a patient lost their beta cells. It is whether their immune system is, right now, capable of mounting an attack that would destroy a transplanted islet. That is a functional question, not a diagnostic one. A patient whose autoimmunity never existed and a patient whose autoimmunity has waned below the level of doing harm are, at the moment of transplant, in nearly the same position: neither has an active attack to launch, and a new islet would survive. Nearly — because a faded autoimmunity is not a vanished one, a distinction the next section has to take seriously.
We can extend this further by noting that many people develop “operational tolerance” to tissues that might have otherwise provoked an immune response. The medical literature is replete with cases where organ transplant patients terminate their immunosuppressants (largely due to the side effects becoming too burdensome) and yet, their organs remain functioning. There’s evidence of this in T1D islet transplant patients too.
I call this group functional T1B, or f-T1B. That is, they may well have no autoimmune activity at all, or they may have a low, weakened immunity, or may be likely to develop operational tolerance.
The label is deliberately functional rather than diagnostic: it gathers everyone whose islet autoimmunity is operationally absent at the time of transplant, however they arrived there. Idiopathic T1B belongs to it. So do the non-autoimmune secondary forms above. And — as the next section will argue — so do some patients formally diagnosed as T1A, whose autoimmunity has worn down over time.
When we take this spectrum into account, it can help shape how the technologies evolve and are applied. Identify the easiest cases first, and build the simplest technology necessary to “cure” them. That is the foundation layer: the patients whose transplant problem contains the fewest immune variables. If the cure cannot be made to work there, it is unlikely to work higher up the structure. If it does work there, every failure and every success tells us what the next layer requires. And people are cured along the way.
From here on, f-T1B means this functional group. The formal idiopathic subtype, Type 1B, I’ll name in full on the few occasions I need it on its own.
Determining Who is “f-T1B”
To determine whether someone is an f-T1B, we need an assay for it. The standard test looks for autoantibodies — but antibodies tell you that autoimmunity has been present, not whether it is still actively attacking. It may also be that the antibodies have waned, but the T cells are still at the ready to attack.
There are assays that can do so, but they are largely in research labs, not yet capable of scaling up for commercial use, but that is beginning to change. A new generation of tests — BASTA (bioRxiv preprint) among them — can detect the beta-cell-specific T cells that do the actual damage, directly, and at a scale a clinic could use. Better still, these assays do not return a yes or a no. They return a magnitude — how much autoreactivity, and against how many targets.
Autoimmunity is not a switch but a throttle.
That reframes eligibility. If autoreactivity comes in degrees, then somewhere along the range is a level below which a graft survives and above which it is destroyed — a threshold, not a category. The question stops being “is this patient autoimmune?” and becomes “is this patient’s autoimmunity below the line where a graft can live without immunosuppression?”
Testing for a person’s degree of immunity in this fashion has precedent, and it’s been in plain view since 2008. A Belgian group (Huurman et al., with Roep and Pipeleers) ran a trial involving 21 established T1D patients — all T1A — where each received cadaveric islets through the portal vein under full immunosuppression. Standard protocol; nothing about the technique was new.
What was new, and unexpected even to the investigators, was a correlation they found after the fact: of the patients with no pre-existing T-cell autoreactivity, seven of eight became insulin-independent; of the four reactive to both GAD and IA-2, none did. Antibody levels — the markers we actually use to sort T1A from T1B — showed no association with outcome at all.
That is not a finding about transplant technique. It is a finding about patient heterogeneity: the T1A label conceals a sizable subset whose autoimmunity is functionally absent. Operative T-cell autoreactivity predicted who kept the graft; antibody status predicted nothing. That is the throttle, measured — operative autoimmunity varying patient to patient among people who all carry the same diagnosis.
That this has sat in the literature since 2008 reveals more about how much more there is in the literature that we can learn from—and act upon. If clinical trials for a given technique show efficacy for patients with particular autoimmune profiles, then the assay can be used to screen them. As the tech improves, the screening score moves with it.
In other words, the architectural plans on how to build the pyramid has already been done. We just weren’t looking at the blueprints.
With that, we have a working definition of the population and a way to identify them.
Now, given the effort, time and expense for pursuing this theory, how do we know the pyramid of recipients is big enough to justify the time, expense and effort? I mentioned that 22% of T1Ds are non-autoimmune, so let’s break down where those numbers come from.
T1B by the Numbers
Assessing the potential number of candidates that might qualify as f-T1B for immunosuppression-free islet transplantation is not yet possible because such a definition never even existed till now.
Instead, we can look at conventional T1B status that’s a subset of f-T1B. Even by conventional measurements, the figures surprise most people. Besides, our primary goal is not to get precise figures, but to validate that the population size justifies the effort, especially if the aggregate complexity, cost and ultimate deployment of this base-case “cure” are markedly improved over the “cure everyone” approach the field is currently pursuing.
This is what funding organizations will be looking for.
We begin with two anchors that we know are solid. For total prevalence, the IDF Diabetes Atlas, 11th Edition / T1D Index v3.0 (2025) estimates 9.5 million people living with T1D globally — up 13% from 8.4 million in 2021. For autoantibody prevalence, the most comprehensive synthesis is a 2022 global scoping review by Ross and colleagues at the Harvard T.H. Chan School of Public Health, which pooled 125 studies across 48 countries and found that islet-autoantibody positivity varies enormously by region — with IA-2A prevalence, for example, ranging from roughly 75% in Europe down to about 31% in Africa.
Overlaying autoantibody-negativity rates onto regional T1D totals produces a rough global picture. The percentages below are estimates — directionally meaningful, not precise — but they make the central point unmistakable: non-autoimmune T1D is not a rounding error, and it is far more concentrated outside of European-ancestry populations.
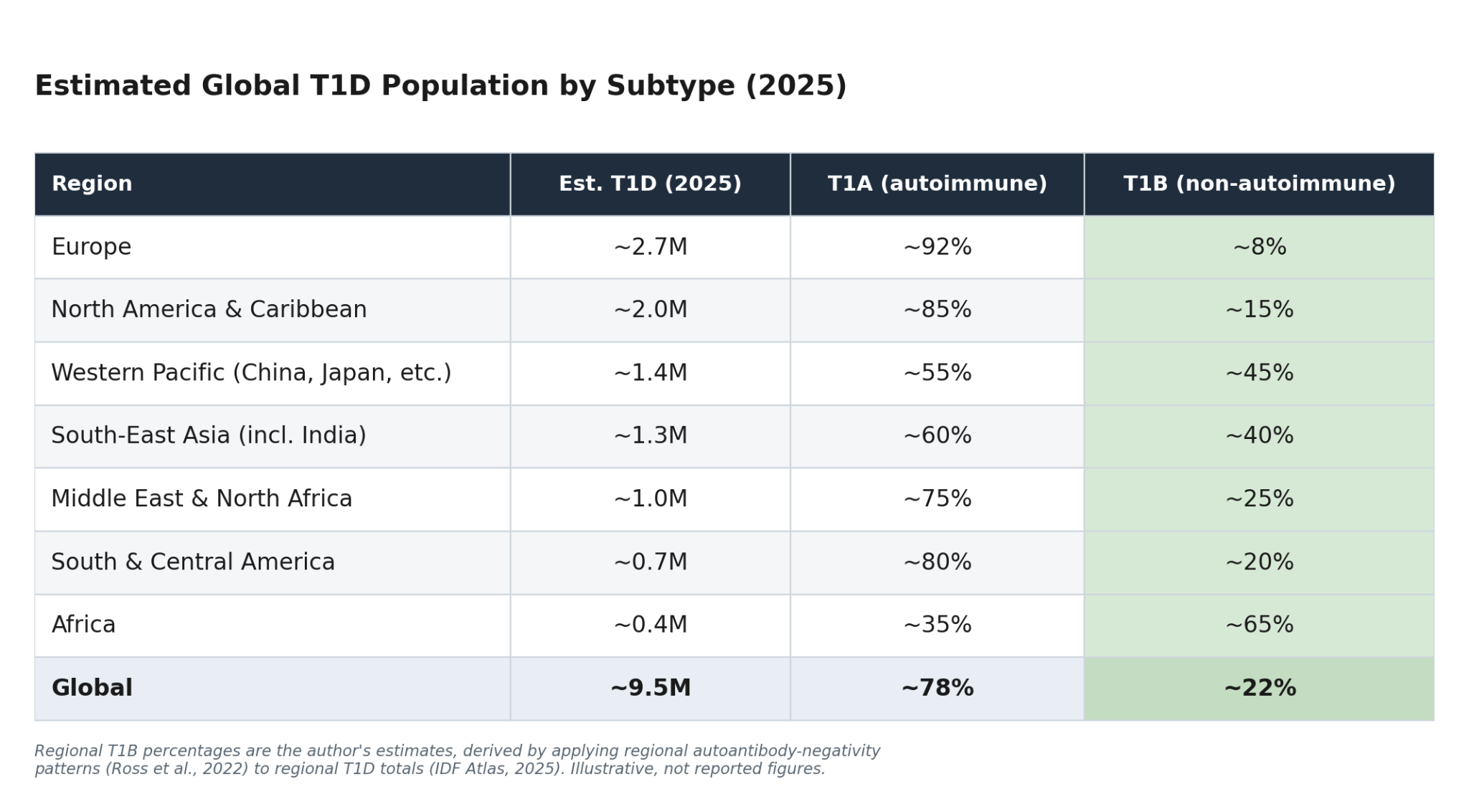
On these estimates, roughly 2 million people worldwide are living with non-autoimmune T1D — and that’s only the fraction we’d capture with today’s incomplete testing. To put 2 million in context, the IDF ranks the largest national T1D populations, in order, as the U.S., India, China, Brazil, the U.K., Germany, Russia, and Canada.
The regional skew is the part worth sitting with. The European and North American figures — the populations most T1D research is built on — are exactly where T1B is rarest. Step outside those populations and the picture inverts. A 2025 study in The Lancet Diabetes & Endocrinology — the Young-Onset Diabetes in sub-Saharan Africa (YODA) study, run across Cameroon, Uganda, and South Africa — found that roughly 65% of young-onset, insulin-treated, clinically diagnosed T1D patients of Black African ancestry were autoantibody-negative, and, crucially, also had a low genetic risk score for autoimmune T1D.
The same pattern, attenuated, shows up in the United States. Comparing the African cohorts against the U.S. SEARCH for Diabetes in Youth data, the researchers found that about 15% of Black Americans diagnosed with T1D showed the same non-autoimmune signature — antibody-negative and low genetic risk.
Put the demographics together and we see that non-autoimmune T1D isn’t a curiosity at the margins. It’s a large, real, and globally uneven population. And it’s highly likely to be undercounted.
Now add the f-T1Bs to the candidate list and the total population could be millions more, and that makes the review article’s premise of autologous SC-islets even more interesting: could we actually cure this population?
We don’t know, because it’s never been tried. But while we consider that, there’s another approach also cited in the review article: hypoimmune islets. These are gene-edited cell lines designed to evade the immune system entirely. Odd as it may sound, the two serve different purposes and have trade-offs between them. So, let’s review both.
Autologous vs. Hypoimmune Islets
Autologous and hypoimmune islets are often discussed as competing technologies, but in this framework they sit at different levels of the pyramid. Autologous islets for f-T1B test the foundation case: can a patient’s own manufactured islets survive when the immune problem is minimised? Hypoimmune islets aim at one or two levels up the pyramid: can one engineered product survive across many immune layers?
Since autologous islets are grown from the patient’s own cells, they carry the patient’s own surface markers, fully exposed to whatever autoreactivity exists. If we’re considering true T1Bs who have no autoimmunity at all, one assumes that there’s nothing to reject, no autoimmunity to reignite, no edits, no device, no drugs. Nothing triggers the immune system.
But is it even possible to grow such cells that are clean of autoimmune response? And will they actually yield glycemic control?
The ADA review article cited two trials that grew autologous SC-islets and put them into patients. One is from Wang and colleagues, from Hongkui Deng’s group. In their paper published in Cell 187, 6152–6164 (2024), Transplantation of chemically induced pluripotent stem-cell-derived islets under abdominal anterior rectus sheath in a type 1 diabetes patient, autologous chemically-induced pluripotent stem-cell-derived islets were transplanted back into the same patient, achieved insulin independence at ~75 days post-transplant, A1c dropped from 7.6% to ~5% range, TIR >98% at one-year follow-up.
The same patient’s own stem cells were used to make new islets, and achieved ideal glycemic control. That alone is worth a moment to ponder: Stem-cell grown islets from the patient’s own tissues physically worked. What we don’t know is if this could be deployed to f-T1Bs because the trial didn’t test for that. In fact, the patient was on immunosuppressants because of two liver transplants for cryptogenic cirrhosis and a whole-pancreas transplant that failed from graft thrombosis.
It’d be nice to see a follow-up trial on a T1B, of course. But we do know there would be at least one main hurdle: the manufacturing process itself. That is, even though the researchers created new islets from the patient’s own tissues, the manufacturing process might have added non-self proteins.
This is exactly the thesis of a 2019 paper from Deuse, Hu, Agbor-Enoh and colleagues (Nature Biotechnology) that showed that reprogramming an ordinary cell into a stem cell, then expanding and maturing it back into an islet cell, accumulates mutations in its mitochondrial DNA (or, mtDNA) — the small, separate genome inside the cell’s energy factories — and some of those mutations make new proteins the patient’s immune system has never encountered. These are neoantigens, which are new molecular flags for the immune system.
In other words, the manufacturing process itself made the product partly foreign. So even a perfectly autologous islet is not immunologically silent — it is “self” at the level of sourcing but not-quite-self at the level of the finished protein.
However, there has yet to be a trial to test the Deuse assertions. What we’d be looking for include:
The effect size in humans. The mechanism and the measurable immune response are real, but no one has shown this is what killed grafts in earlier trials, such as Deng’s group, where immunosuppression was used.
The magnitude next to autoimmune recurrence. A handful of new flags is a bounded, tunable problem — not the primed immune army that has hunted beta cells for decades.
Mitigation and whether it’s tractable. The mutation load tracks with how the cells were reprogrammed, how long they were grown in culture, and the donor’s age; cleaner, shorter manufacturing lowers it. This is engineering, not biology.
Whether an SC-cell line can be screened for these mutations before use — though more of them accumulate as the cells mature into islets, after the screening point, so screening contains the problem rather than erasing it.
These would tell us how much further work is needed, if any.
This is what brings us to an alternative to autologous islets, which happens to be where the field is moving already: hypoimmune islets. This is a single cell line, rather than a per-person line, where the cells are gene-edited to “hide” from the immune system by stripping the surface flags (MHC) that let T cells inspect what’s inside. (If they can’t see inside, they don’t know that it’s a foreign body that needs to be attacked.) Actually, it’s two-for-one against allo and auto (because both are MHC-restricted), so the idea is that these hypoimmune islets would be built once and produced for everyone — drug-free because the edits do the foreign-tissue evasion that the autologous route accomplishes. It could/should work for both the T1A and T1B populations.
A demonstration of the gene edits can be seen in Sana’s UP421, which involves hypoimmune-edited islets placed in a long-standing T1A patient with no immunosuppression. After fourteen months, the islets were still producing insulin and surviving.
But, like the Deng group’s trial, UP421 just missed telling us what we wanted to know because they didn’t use SC-islets — they edited cadaveric islets (just to isolate the gene editing aspect). In effect, neither trial gets us what we want to know:
In the Deng group, they used autologous stem-cell grown islets along with immunosuppression.
In UP421, they didn’t use immunosuppressants, but they also didn’t use stem-cell islets; they used cadaveric donor islets (which they edited).
What Sana would need to do next is do the same edit to SC-islets from their chosen cell line. And this is where those carry a broader exposure.
Yes, their edits work because they strip off the molecular ID badges — the MHC proteins — that every cell shows so the immune system can inspect it, so a patrolling immune cell finds nothing to read and moves on. But the immune system can still poison the islets from a distance with inflammatory signals (the cytokine arm); and because one cell line would be used for everyone, antibodies can latch onto leftover surface proteins and call in complement, an antibody-driven (humoral) attack the badge-stripping never touches.
There’s even a twist the edits create rather than fix: natural killer cells are built to destroy any cell that isn’t showing an ID badge — a trigger known as “missing self” — so removing the badge makes them more suspicious, not less. Anticipating that, the designers answer with yet another edit — a “don’t-eat-me” signal called CD47.
And that’s the pattern in miniature: each fix closes one door and tends to open the next. Lather. Rinse. Repeat.
Whether the whole stack holds up inside a living human immune system is exactly what no one has shown.
These and other concerns make hypoimmune islets just as much a thorny theoretical approach as autologous islets. If they work, they’ll take care of the autologous SC-islet track, and T1A and T1B can be cured by the same technology. Unless and until they do, the autologous SC-islets are easier, less complicated, and can serve a substantial population right away.
This leads directly into pragmatism. In other words, money.
An Economic Consideration
Clinical trials are expensive. Measured against the trials the field actually runs, an autologous-T1B pilot would be far leaner. For comparison:
Organ acquisition alone for a single patient runs $50,000–90,000, because each patient typically needs islets from 2–3 deceased-donor pancreata. Counting pancreata that are processed but fail release criteria, this rises to as much as $180,000 per patient.
A U.S. analysis put the fully-loaded cost of islet transplantation alone (ITA) at $138,872 (Moassesfar et al., American Journal of Transplantation, 2016). Those per-patient costs are dominated by the two cost centers an autologous-T1B trial removes — organ acquisition and the immunosuppression management donor cells require.
A trial testing a new immunosuppression regimen carries that donor cost plus the drug arm and the extended safety monitoring that immunosuppression demands.
A trial testing an encapsulation device carries the donor cost plus the device.
Because of their complexity, these trials cost vastly more than what an autologous SC-islet T1B trial would by stripping those variables out: no donor organs to procure (the cells are the patient’s own), no immunosuppression arm (there’s no immunosuppression), no device (the cells go in unencapsulated).
What remains is a structurally simpler trial — fewer variables, cleaner monitoring, healthier subjects, and none of the multi-donor organ logistics that have constrained islet trials for thirty years. For scale, the NIH’s Clinical Islet Transplantation Consortium ran its Phase 2/3 protocols across roughly 100 patients at multiple centers; a T1B pilot needs only 10–15 to establish baseline feasibility before any comparative trial.
That covers the trial. The larger expense sits upstream in the R&D and infrastructure needed to have a cell product to go to trial in the first place. Consider that Vertex paid $950 million to acquire Semma Therapeutics in 2019 and an additional $320 million to acquire ViaCyte in 2022 — $1.27 billion in acquisitions alone, before the years of internal R&D and manufacturing buildout that followed. Vertex’s three parallel programs all exist to solve one problem: getting cells past a host immune system that wants to destroy them. VX-880/zimislecel does it with lifelong immunosuppression, VX-264 by sealing the cells in an encapsulation device, and the hypoimmune line by gene-editing the cells to hide.
Compare all that to the easier autologous. The patient’s own cells, in a host that isn’t attacking them, and that requires no drugs, no devices, and no edits.
Autologous iPSC-derived islet manufacturing is feasible today. Deng’s group in Tianjin has produced clinical-grade autologous islets for individual patients. And yes, it’s nascent and expensive — roughly $100K per patient in cell production alone, before surgery and follow-up.
Scalability is a potential yellow flag of course, but the alternative is hypoimmune SC-islets. While the hypoimmune, single cell line could scale better from a manufacturing perspective, it has a much harder uphill road for technically working, especially as the target patient goes beyond the T1B population. This doesn’t make it worse, nor does it imply that it’s one or the other. But it is definitely too soon to pretend we have a crystal ball on either of these concerns to make confident forecasts.
Nevertheless, the benefit of treating the easiest patients first allows for an acceleration of the process in ways that have both scientific and economic benefits that the structural advantages (cheaper, cleaner, faster) start accruing from day one relative to the comparators, and can benefit millions of people.
The economics are so favorable, and the potential result so widely beneficial, that the low barrier to entry makes this an attractive value proposition for a small startup to bring to investors, funding organizations, and granting agencies.
Building from the Ground Up
This is where we loop back to the ADA review we began with. Its authors are right that the immune system is the next frontier, and right to lay out the four ways across it. What the ground-up approach adds isn’t a fifth way across — it’s the map.
Each of those four is being engineered for one abstract enemy: the maximally hostile immune system, imagined rather than measured. The ground-up approach measures it, and the foundational trials it produces are exactly what will benefit those other technologies.
Begin with the patient who has no immune fight at all—non-autoimmune—no door is needed, just a refinement of the basic autologous SC-islet manufacturing process. Insert enough autologous islet mass to test for an immune response, not for glycemic control. If the graft draws an immune response because of that neoantigen the Deuse paper suggests might happen, experiment with some of the new milder immunosuppressants at low doses. Eledon’s tegoprubart comes to mind. Low dose, low duration.
As the immune response arm is addressed, also test for glycemic control by increasing islet mass and experimenting with different site locations — the anterior rectus sheath and the omentum are currently interesting candidates.
As results progress, recruit f-T1Bs with low grade autoimmunity. As the autoimmune signal climbs, the data says what the minimum sufficient answer is — a gene edit, a nudge toward tolerance, a slighting longer duration of immunosuppressants. Nothing toxic and not life-long.
As we iterate through the various degrees of autoimmunity, we may eventually find that we’ve cured a large share of the T1D population, leaving only the most acutely affected for last. These are the people where the most complex and expensive interventions might have to be used.
You know, the interventions that are being developed today with billions of dollars. It’s a pyramid scheme gone awry.
We do not have to decide in advance which technology will cure everyone. We only have to stop trying to place the capstone first. Start at the foundation: the patient whose immune system poses the least resistance. Learn what works there. Then build upward, one immune layer at a time.
Thank you very much for the article. So, I am one of these anomalies, and I was tired of proving to doctors that cases like mine exist. I estimate my 'autoimmune event' occurred 17 years ago, but even in my childhood, there were signs of low beta-cell activity (fatigue, constantly blue extremities).
Anyway... I frame health and illness within a framework of energy availability and I abolish rigid categories in metabolism. The cell doesn't care whether it is sitting in front of an overfull or a half-empty plate. If nothing arrives, nothing arrives. I would also broaden the scope much, much further, but that is speculation: how do you see this? I believe that for many of the conditions involving extreme chronic fatigue that I won't name here... could it be an insulin-timing problem?
Through stoic serenity and a doctor who was willing to look at physiology rather than ICD codes, I managed to secure basal insulin for myself, and I still run this protocol at my own discretion and according to my own rhythms. This goes hand-in-hand with a "restricted" diet and regular movement several times a day, which poses no problem for me because I have the routine down.
As a 4 year LADA T1d I am very strict with my diet but my insulin resistance is not improving. In fact, it seems to be going in the wrong direction. I am so tired of the anxiety caused by my Dexcom alarms that I’m high or low. Currently, I am taking about 25-30 units of insulin a day- I am not overweight - and walk daily! Any input or advice from anyone is appreciated!